![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

The purpose of this practical is to become familiar with the use of the ClustalX program. This program is written with the assistance of the NCBI toolbox for software development. This means that the program runs on all operating systems with a common graphical user interface (GUI) on each system.
Depending on the operating system, you will start the program either by double-clicking the ClustalX icon or by typing 'clustalx' (no parentheses) at the command line (UNIX).
The first screen that you will see will look like this:
You will notice at the top, left hand side of the screen is a pull-down menu. Clicking on this menu allows you to change between the two major modes of alignment of ClustalX:
In order to load some sequences into memory, you must select the file menu with the mouse:

If you select the option to load sequences, they will be displayed in
the main window. ClustalX accepts
7
formats at present.

If, on the other hand, you wish to align two existing alignments together or to align a single sequence to a pre-existing alignment, then you can switch to profile alignment mode. In this case, the screen is split. when you have two profiles loaded, the screen will look something like this:
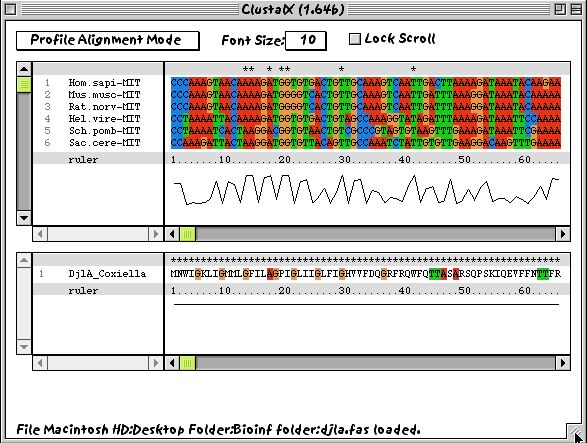
If you wish to manipulate sequences, for example, re-arranging the order of the sequences in the window, removing gaps, removing columns of data that contain only gaps (this can sometimes happen when you remove sequences, the remaining sequences have columns with gaps only), you can do so using the commands available under the edit menu.

Perhaps the most important menu is the one dealing with alignments. Under this menu, it is possible to change the alignment parameters, both for pairwise alignment and for the multiple alignment stages. If a profile alignment is being carried out, or if sequences with gap characters are being used in the alignment process, then it is possible to remove the gap characters prior to alignment.
A complete alignment may be carried out, or alternatively, the first
stage of the alignment process (producing the guide tree from the initial
pairwise alignments) may be carried out, or even an old guide tree may
be used as the starting point for the alignment process, therby eliminating
the need for the pairwise alignments.
ClustalX may also be used in order to infer the relationships between sequences. This part of the program should be treated with extreme caution. The only alignments that should be used are those where positional homology is guaranteed. Unfortunately, automated alignment can be prone to error and uncertainty.
However, given a reliable alignment, where every position is aligned with absolute certainty to its homologs in the other sequences, ClustalX can be very useful for drawing phylogenetic trees. There are some basic corrections for multiple substitutions and the reliability of the resulting relationships can be estimated by bootstrapping.
However, it is our belief that the most important features of ClustalX are not in its tree-drawing capabilities and that generating phylogenetic hypotheses is best done using other products.


One of the interesting advantages of using ClustalX over ClustalW is the ability to visually evaluate the quality of the alignment. In particular, the ability to highlight areas where the alignment is poor is a significant advantage. Is is possible that one sequence has a short region that shows low levels of residue similarity to the rest of the alignment. This can be due to evolutionary processes, or quite frequently, due to errors in sequencing. A frameshift error that is corrected by a frameshift downstream might go unnoticed and become submitted to GenBank. ClustalX is capable of detecting these kinds of errors. An example of the utility of this method is shown:
Here is a typical alignment:
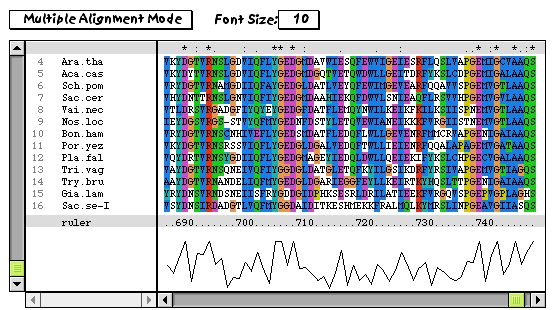
However, choosing a segment length of 7 residues and using the Gonnet 250 matrix, the program can choose regions of the alignment that are showing exceptionally low scores.
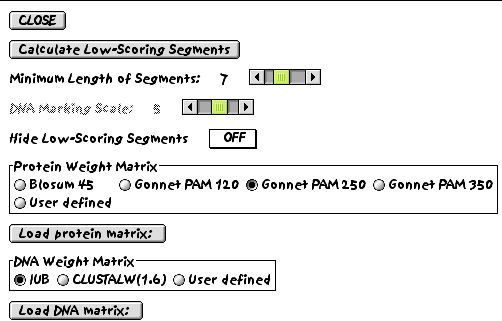
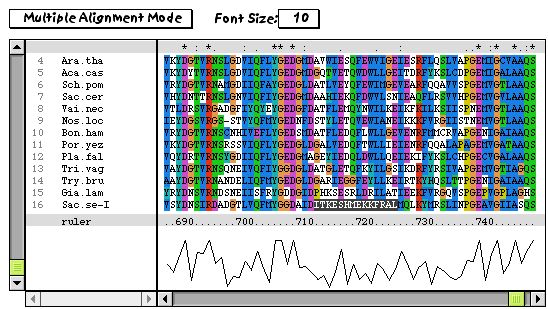
The bottom sequence has a region of 14 amino acid residues that show a surprising amount of dissimilarity compared to the others (note the darkly-shaded residues). It is possible that either this region of exceptional divergence is due to misalignment, relaxed selective pressures on this region, or due to sequencing errors.
In addition to all of the other features, ClustalX offers a short description of the available commands, using the help menu. You can select any of these options for a brief description of their usage.

This brief description is not intended to replace the ClustalX information page. Please visit this page and read its contents.
You can now proceed to today's Exercise.
<<Back | Next >>