


| |
|
|
| http://www.dbbm.fiocruz.br/labwim/bioinfoteam/ | bioinfoteam@fiocruz.br |
The widely used programs BLAST (both National Center for Biotechnology Information [NCBI] and Washington University versions) and FASTA for similarity searches in nucleotide and protein databases usually result in copious output. However, when large query sets are used, human inspection rapidly becomes impractical. BioParser is a Perl program for parsing sequence similarity analysis reports. Making extensive use of the BioPerl Toolkit, the program filters, stores and returns components of these reports in either ASCII or HTML format. BioParser is also capable of automatically feeding a local MySQL database with the parsed information, allowing subsequent filtering of hits and/or alignments with specific attributes.
The new version is able to parse and analyze the results obtained with the sequence similarity search program HMMER (both HMMSEARCH and HMMPFAM). In addition, the BioParser Browser was improved with new search fields (QDesc, HDesc, QLength, HLength) and operators (like/not like, and/or), making BioParser even more flexible. Request a copy.
An on-line version of BioParser is now available. Parse and analyze your BLAST, FASTA, SSEARCH or HMMER result in our server freely!
BioParser 1.2.0 has been updated with the Concurrent Versions System (CVS) of BioPerl. The current and past distributions of BioPerl are UNABLE to handle the new BLAST output style.
BioParser has been updated with version 1.5.2 of BioPerl. The new BioPerl version introduces some useful features such as Bio::SearchIO speed up, and it contains many bug fixes since the 1.5.1 release;
The function accounting for the Run SQL field has been updated to avoid misinterpretation of some mathematical symbols;
Miscalculations of "Queries without Hits" and "Hits without HSPs" reports have been fixed;
Other small changes in the code have been made to improve the way BioParser deals with the sequence frame information in TBLASTN, TFASTX, and TFASTY reports, and also to accurately calculate the Ident(%) and Pos(%) in HMMSEARCH and HMMPFAM reports.

Some numeric data fields in the BioParser MySQL database structure has been updated to account for sequence similarity reports containing huge number of queries/hits/HSPs and/or huge-sized sequences.
BioParser v1.2.3
This software is licensed for non-commercial use only.
Distributed under the terms of:
Creative Commons Attribution-NonCommercial-NoDerivs 2.0 License.
http://creativecommons.org/licenses/by-nc-nd/2.0/legalcode
BioParser needs only one working account to work with the database, but it can also be set up with two different working accounts (read-write/read-only) as well. For security purposes, for instance, the parser may be given a read-write account while the CGI interface may be given just a read-only account. To create the default BioParser database, fire up the MySQL client (mysql under Unix or mysql.exe under Windows), and issue the following command:
This creates a database to import the data; you can choose any name you wish. Now you need to "enter" your database with this command:
To create the default tables, fire up the mysql client and issue the following command:
NOTE: Under windows you can use both "/" or "\" to delimit directories. Importantly, the directory cannot contain spaces, otherwise, mysql may not even find the file. The default tables created by bioparser.sql are: bp_query, bp_hit, bp_hsp and bp_report (Figure 10). You can change the name of the tables running the following command:
BioParser depends on the following softwares:
NOTE: MySQL 5 is not supported yet.
Perl - http://www.perl.org/
ActiveState Perl for Windows - http://www.activestate.com/Products/ActivePerl/
MySQL Database - http://dev.mysql.com/downloads/mysql/4.0.html
You also need the following Perl Modules:
Unix users can find Perl Modules in the Comprehensive Perl Archive Network (CPAN) website at http://search.cpan.org/.
Windows users, using ActiveState Perl, should use the "ppm" utility to search and install the required modules.
It's highly recomended that you use the provided BioPerl distribution (version 1.5.2) found in the "bioperl/unix" or "bioperl/windows/" directory. The new BioPerl version introduces some useful features such as Bio::SearchIO speed up, and it contains many bug fixes since the 1.5.1 release. Windows users can install it using the command:
You need to set up both the parser (BioParser GUI) and CGI (BioParser CGI browser) interfaces. The parser has a built-in configuration in its GUI to input the database information. The CGI interface can be set up with a regular text editor. Both programs use xml files to store their config, which is pretty straightforward.
To set up the CGI, edit the config.xml file in the "browser" directory, which has the following structure:
You also need to set up the available databases by editing the dbase.xml file located in the "browser" directory as follow:
Installation is pretty simple, you just need to copy the contents of some directories found in the BioParser distribution to a new directory of your choice.
For the BioParser GUI, copy the contents of the "parser" directory to wherever you want to host those files (eg.: /usr/local/bioparser).
For the BioParser CGI browser, copy the contents of the "browser" directory to your web hosting directory (eg.: ~/public_html/bioparser/).
To work with BioParser browser, launch your web browser and type:
If you have followed each step correctly, you should see the main query page.
NOTE: You need a CGI-enabled web-server (preferably Apache) with perl and the required modules installed.
BioParser accepts any BLAST (BLASTN, BLASTP, BLASTX, TBLASTN, TBLASTX) [NCBI or Washington University version], FASTA (FASTA, FASTX/FASTY, TFASTX/TFASTY), SSEARCH or HMMER (HMMSEARCH, HMMPFAM) output in ASCII (plain text) or XML (supported only for NCBI BLAST version) format. The program does not support any other input format. It parses single or multiple reports, i.e., searching results from one or several query sequences (or profiles) simultaneously. The files single_blast_report and multiple_blast_report are examples of BLASTP run outputs (ASCII format) for a single query and for multiple (three in this example) query sequences, respectively. Before parsing your sequence alignment report, be sure that it follows exactly one of these accepted formats so as to avoid BioParser crashing.
A schematic representation of the full system architecture is presented in Figure 1. Basically, BioParser takes a BLAST, FASTA, SSEARCH or HMMER report file as an input and uses the Bio::SearchIO module of the BioPerl library to parse most of the information in this file.
Three different parsing options are offered: saving the parsed information in (i) ASCII (plain text) or (ii) HTML format, in which the parsed elements are displayed as a table (columns representing the sequence similarity report attributes, and lines corresponding to distinct alignments) or a list (with the following nested structure: results -> query -> hit -> HSP; see Table 1), respectively, or (iii) transferring the parsed information to a local MySQL database, which is done automatically by BioParser. This last option is the preferred parsing option for the analysis of large data sets.
BioParser also includes a web-based interface (BioParser Browser), which offers a user-friendly environment to interact with the MySQL database, allowing the user to apply a number of selection criteria to the parsed data so as to filter out hits and/or alignments with specific features (see section Filtering Options and Usage).
Table 1 summarizes the available parsing options and all BLAST, FASTA, SSEARCH or HMMER attributes which can be parsed with BioParser.
Examples of ASCII and HTML outputs can be found in the BioParser Output Files section of this page.
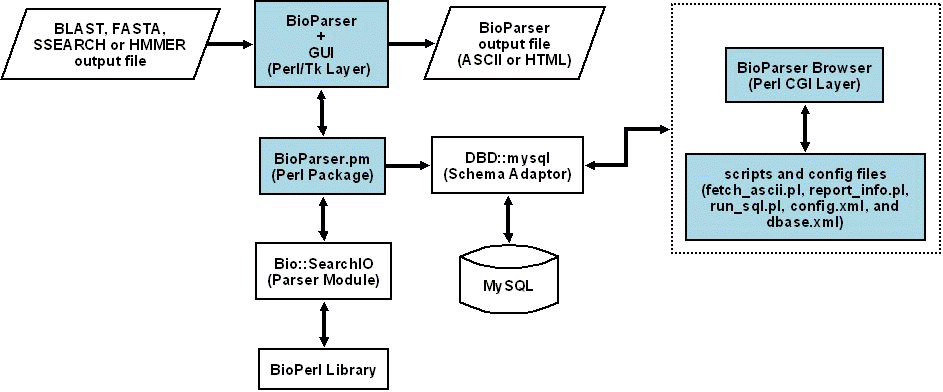
Figure 1. Schematic representation of the BioParser system architecture.
| Parsing Options and Attributes | ||||
| Report Section | HTML | ASCII | Database | Description |
| RESULTS | Algorithm | Algorithm | algorithm | Algorithm |
| RESULTS | Version | Version | version | Algorithm version |
| RESULTS | DB Name | DB Name | db_name | Database name |
| RESULTS | DB Letters | DB Letters | db_letters | Number of residues in database |
| RESULTS | DB Entries | DB Entries | db_entries | Number of database entries |
| RESULTS | Parameters | Parameters | parameters | Parameters used |
| QUERY | Name | QueryName | query_name | Query name |
| QUERY | Accession | - | query_accession | Query accession number |
| QUERY | Description | - | query_desc | Query description |
| QUERY | Length | QLength | query_len | Query length |
| QUERY | No. Hits | - | - | Number of hits |
| HIT | Name | HitName | hit_name | Hit name |
| HIT | Accession | - | hit_accession | Hit accession number |
| HIT | Description | - | hit_desc | Hit description |
| HIT | Length | HLength | hit_len | Hit length |
| HIT | Identities (%) | HIdent(%) | hit_fr_identities | Overall fraction of identical positions across all HSPs (aligned regions only) |
| HIT | Positives (%) | HPos(%) | hit_fr_positives | Overall fraction of conserved positions across all HSPs (aligned regions only) |
| HIT | Frac. Aligned Query (%) | HAlnQuery(%) | hit_fr_aln_query | Fraction of the query sequence which has been aligned across all HSPs (not including intervals between non-overlapping HSPs) |
| HIT | Frac. Aligned Hit (%) | HAlnHit(%) | hit_fr_aln_hit | Fraction of the hit sequence which has been aligned across all HSPs (not including intervals between non-overlapping HSPs) |
| HIT | No. HSPs | - | - | Number of HSPs for a given hit |
| HSP | HSP Rank | HSP | - | Rank of the HSP within a given hit |
| HSP | Query Frame | QFrame | hsp_query_frame | Frame of the query sequence (0 = -1/+1; 1 = -2/+2; 2 = -3/+3); not defined for (wu-)blastn, (wu-)blastp, fastn, fastp and (wu-)tblastn |
| HSP | Hit Frame | HFrame | hsp_hit_frame | Frame of the hit sequence (0 = -1/+1; 1 = -2/+2; 2 = -3/+3); not defined for (wu-)blastn, (wu-)blastp, fastn, fastp and (wu-)blastx |
| HSP | HSP Query Strand | QStrand | hsp_query_strand | Strand of the query (1 = Plus; -1 = Minus; 0 = not defined) |
| HSP | Hit Strand | HStrand | hsp_hit_strand | Strand of the hit (1 = Plus; -1 = Minus; 0 = not defined) |
| HSP | Score | Score | hsp_score | Raw score |
| HSP | Bits | Bits | hsp_bitscore | Bit score |
| HSP | E value | Evalue | hsp_evalue | Expectation value for the HSP (e-value) |
| HSP | Identities | Ident | hsp_identities | Number of identical residues |
| HSP | Identities (%) | Ident(%) | hsp_fr_identities | Fraction of identical positions for a given HSP |
| HSP | Positives | Pos | hsp_positives | Number of conserved residues |
| HSP | Positives (%) | Pos(%) | hsp_fr_positives | Fraction of conserved positions for a given HSP |
| HSP | Query Gaps | QGaps | hsp_query_gaps | Number of gaps in the query alignment |
| HSP | Hit Gaps | HGaps | hsp_hit_gaps | Number of gaps in the hit alignment |
| HSP | HSP Length | HSPLen | hsp_length | Length of HSP (full length of the alignment) |
| HSP | Query Overlap | QOverlap | hsp_query_overlap | Length of query participating in alignment minus gaps |
| HSP | Hit Overlap | HOverlap | hsp_hit_overlap | Length of hit participating in alignment minus gaps |
| HSP | Frac. Aligned Query (%) | AlnQuery(%) | hsp_fr_aln_query | Fraction of the query sequence which has been aligned within a given HSP |
| HSP | Frac. Aligned Hit (%) | AlnHit(%) | hsp_fr_aln_hit | Fraction of the hit sequence which has been aligned within a given HSP |
| HSP | - | - | hsp_query_start | Query start position from the alignment |
| HSP | - | - | hsp_query_end | Query end position from the alignment |
| HSP | - | - | hsp_hit_start | Hit start position from the alignment |
| HSP | - | - | hsp_hit_end | Hit end position from the alignment |
| HSP | Query range | QRange | - | Query start and end positions from the alignment |
| HSP | Hit range | HRange | - | Hit start and end positions from the alignment |
Usage
Parsing to ASCII or HTML format
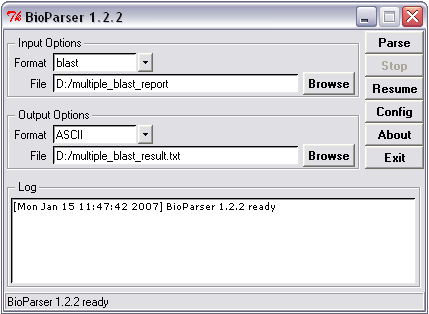
Figure 2. Example of parsing to ASCII format.
Parsing to a local MySQL database
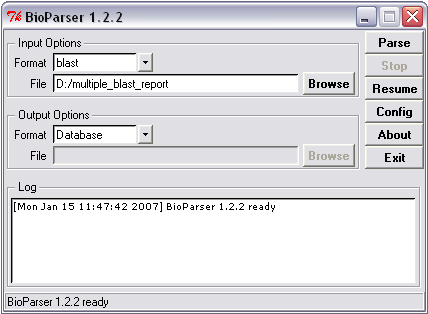
Figure 3. Example of parsing to database.
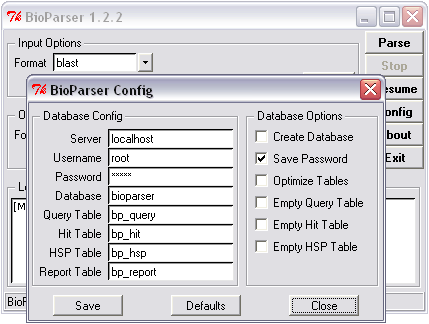
Figure 4. Database configuration.
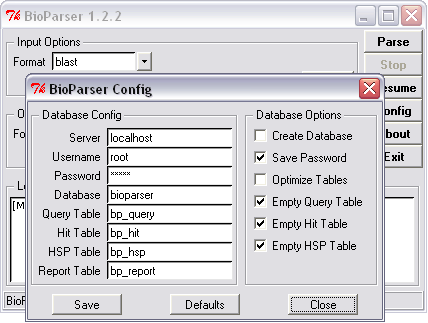
Figure 5. Empting all tables of an existing database before parsing the new information. |
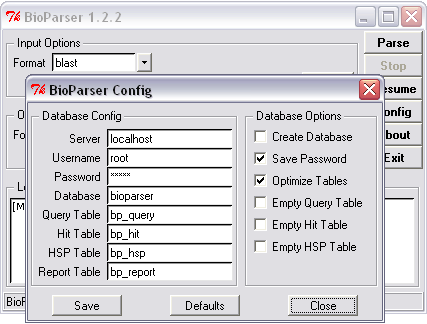
Figure 6. Updating all tables of an existing database with new parsed information. |
BioParser has been empowered with a crash recovery system which permits the interruption of the parsing-to-database process, without data loss. The parsing process can be resumed according to the following instructions:
If the parsed information has been stored in a local MySQL database the user can select queries which are related to hits and/or alignments with particular attributes through the BioParser browser (Figure 7). At least thirteen different attributes can be used to filter out the parsed result: QueryName (name of the query sequence), HitName (name of the hit sequence), QDesc (description of the query sequence), HDesc (description of the hit sequence), QLength (length of the query sequence), HLength (length of the hit sequence), Score, Bits, Ident (%) (fraction of identical positions for a given HSP), AlnQuery(%) (fraction of the query sequence which has been aligned within a given HSP), AlnHit(%) (fraction of the hit sequence which has been aligned within a given HSP), Evalue (expectation value for the HSP), and SizeDiff (difference in length, expressed as a fraction, between the query and hit sequences). Users can choose one or a combination of attributes, connecting them with a logical AND or a logical OR.
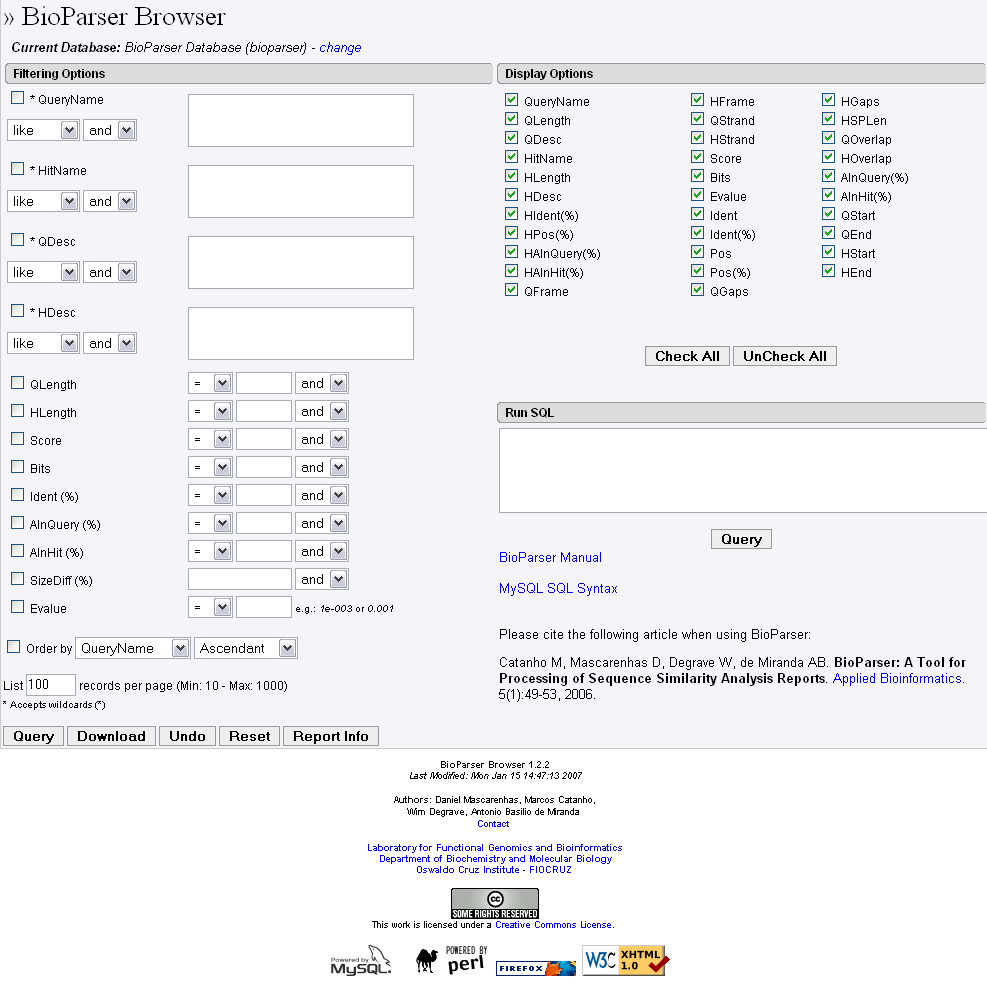
Figure 7. An overview of the BioParser browser showing the filtering and display options, and the SQL field.
Usage
To demonstrate the applications of BioParser browser, the BLASTP output file multiple_blast_report will be used as an example. This file contains reports resulting from consecutive BLAST searches against the NCBI Protein Reference Sequences database (RefSeq) using three distinct hypothetical proteins encoded by the human parasite Trypanosoma cruzi. After parsing the multiple_blast_report file and loading it into a local MySQL database with BioParser, let us select only the records in which the fraction of identical positions in the HSP is greater than or equal to 90% AND the fraction of the query sequence which has been aligned within the HSP is greater than or equal to 90% AND the difference in length between the query and hit sequences is within 20% (for example, if a query sequence has 100 residues, any hit sequence ranging from 80 residues to 120 residues would satisfy this constraint, and vice-versa). Also, let us select only a few attributes to be displayed in the BioParser browser web output: QueryName, QLength, HitName, HLength, Score, Bits, Evalue, Ident(%), Pos(%), QGaps, HGaps, HSPLen, AlnQuery(%), and AlnHit(%). The procedure is straightforward (Figure 8):
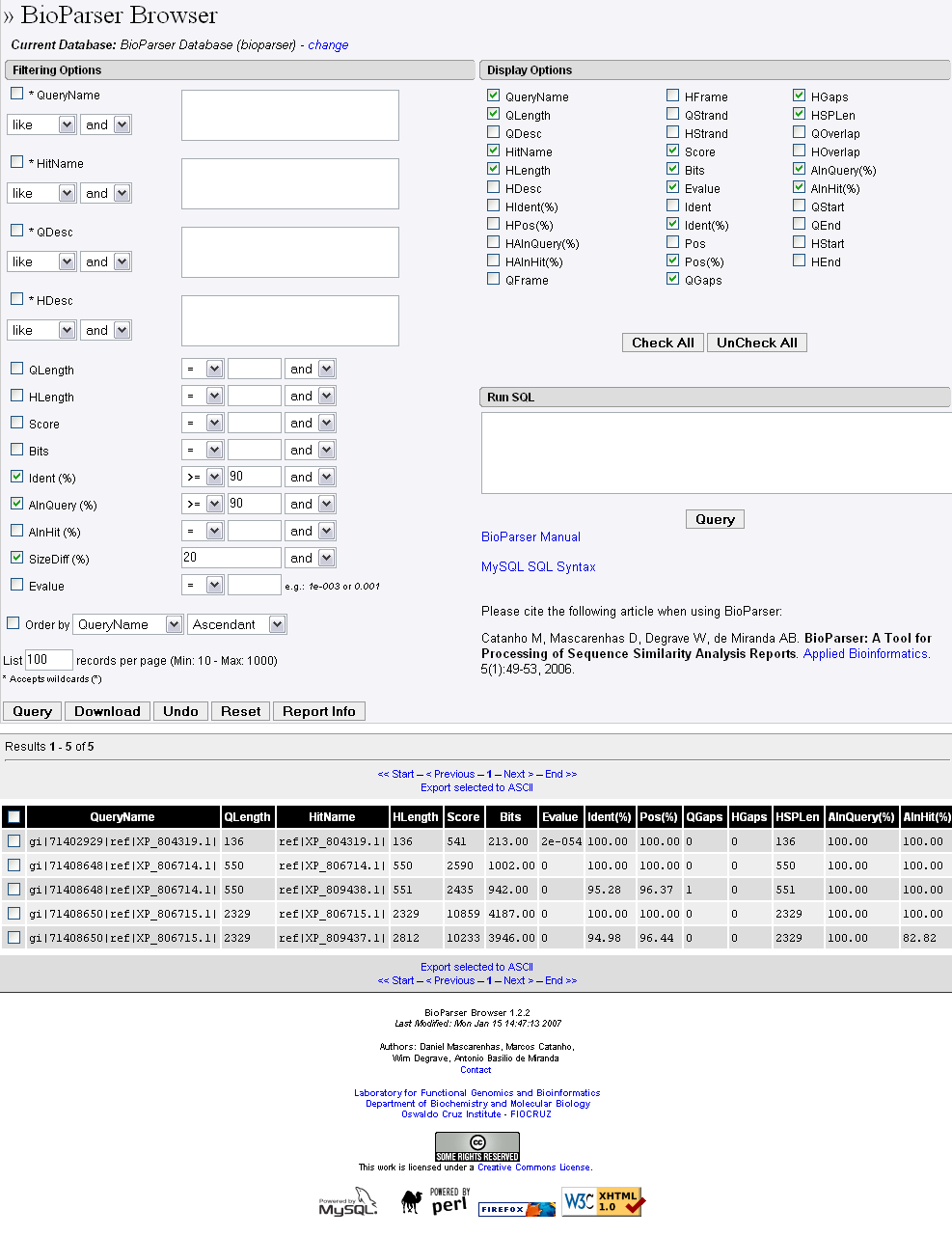
Figure 8. Screenshot of the BioParser browser showing an example of filtering usage and the corresponding database searching result (see text for explanation).
The database searching result is displayed as a table in which the columns represent the selected sequence similarity attributes, and each line corresponds to a different alignment (Figure 8).
In this example, only 5 alignments out of 335 (total number of HSPs presented in the BLASTP report; press the Report Info button for details) satisfy the constraints. Records 1, 2 and 4 (from the top) correspond to self-alignments, whereas records 3 and 5 indicate two possible duplications in the genome of T. cruzi (the sequences in each pair are nearly identical).
Selected records can be exported to a plain text file. Just click check the records you would like to export (or Check All) and click the Export selected to ASCII link (Figure 8). Press the Download button if you want to export all records retrieved to a plain text file in one single step (Figure 8).
More experienced users can refine their selection using the SQL search field (Run SQL) employing Structured Query Language (see the syntax for SQL statements supported in MySQL at http://dev.mysql.com/doc/mysql/en/SQL_Syntax.html). The required information to SQL search the database created by BioParser is displayed in Figure 9 and Figure 10 (the default database structure and its tables and fields, respectively).
The proposed BioParser database structure is simple and intuitive: for each aligned pair present in the sequence similarity report, the attributes related to the query and hit sequences are stored (without redundancy) in the bp_query and bp_hit tables, respectively. The attributes that characterize each alignment (HSP) are stored in the bp_hsp table, which is linked to the query and hit tables by two foreign keys: query_id and hit_id, respectively (Figure 9).
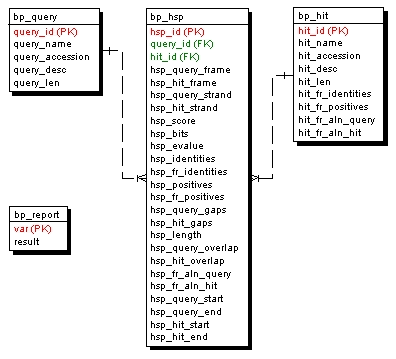
Figure 9. Entity-Relationship Diagram showing the relational structure of the default database created by BioParser. PK - primary key; FK - foreign key.
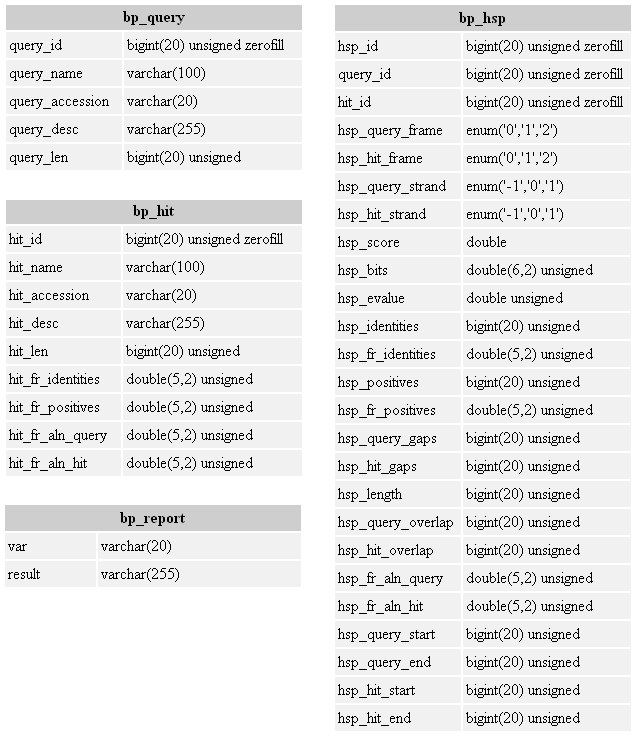
Figure 10. An outline of the BioParser default database tables and fields. The var field in the bp_report table contains the following information: algorithm, version, database (db) name, db letters, db entries, parameters, total queries, total hits, total hsps, queries without hits, and hits without hsps.
The following files are parsed results obtained with the previous BLASTP outputs (single_blast_report and multiple_blast_report) using BioParser:
Please cite the following article when using BioParser:
Catanho M, Mascarenhas D, Degrave W, de Miranda AB. BioParser: A Tool for Processing of Sequence Similarity Analysis Reports. Applied Bioinformatics. 5(1):49-53, 2006. [PubMed] [PDF]
To obtain a copy of BioParser, please contact bioinfoteam@fiocruz.br.
Comments, questions, suggestions and problem reports are also welcome.