
|
| Home: http://www.dbbm.fiocruz.br/labwim/bioinfoteam/ | E-mail: bioinfoteam@fiocruz.br |
Ajuda no mapeamento de relações entre proteínas
O World Community Grid e o Instituto Oswaldo Cruz, Fiocruz, irão comparar informação genômica para melhorar a qualidade e a interpretação de dados biológicos e nosso conhecimento de sistemas biológicos e das interações entre os parasitas, seus hospedeiros e ambiente. Esta informação pode ter um papel crucial no desenvolvimento de melhores drogas e vacinas, assim como melhores métodos de diagnóstico.
Para questões específicas sobre o Projeto de Comparação de Genomas, dirija-se por favor à página de perguntas freqüentes do projeto.
O Projeto de Comparação de Genomas (The Genome Comparison Project): Aperfeiçoando a anotação funcional das proteínas nas bases de dados
Com o passar dos anos, uma grande quantidade de informação secundária (estrutural, funcional, similaridades a outros registros e uma variedade de referências cruzadas) foi adicionada aos registros contendo as seqüências protéicas. Uma vez que tal informação seja adicionada, ela raramente sofre atualizações e/ou correções. Conseqüentemente, a anotação da função protéica predita é freqüentemente incompleta, utiliza nomenclatura não-padronizada ou pode até mesmo ser incorreta, resultado de sua inferência a partir de seqüências previamente analisadas e incorretamente anotadas. Adicionalmente, muitas proteínas são compostas de várias estruturas e/ou domínios funcionais (módulos compreendendo distintas unidades estruturais, funcionais e evolutivas), as quais podem ser ignoradas por procedimentos automáticos de anotação. Além disso, a informação comparativa disponível atualmente é enorme quando comparada aos primeiros dias da genômica.
O objetivo principal do Projeto de Comparação de Genomas é a execução completa de comparações par-a-par entre todas as seqüências protéicas preditas, possibilitando a obtenção de índices de similaridade que serão utilizados, juntamente com uma nomenclatura padronizada de genes e seus produtos, o Gene Ontology (http://www.geneontology.org), na construção de um repositório referência para a comunidade de anotadores, proporcionando uma fonte inestimável de dados para os biólogos. O programa de comparação de seqüências utilizado no Projeto de Comparação de Genomas é chamado SSEARCH (W.R. Pearson [1991] Genomics 11:635-650), uma implementação gratuita e aberta do algoritmo de Smith-Waterman (T. F. Smith and M. S. Waterman, [1981] J. Mol. Biol. 147:195-197), o qual encontra o melhor alinhamento local (do ponto de vista matemático) entre pares de seqüências.
Como resultado do projeto, uma anotação precisa e padronizada,
a correção de inconsistências e a atribuição
de possíveis funções à proteínas hipotéticas
de função desconhecida serão facilitadas. Adicionalmente,
proteínas com múltiplos domínios e elementos funcionais
serão corretamente identificadas. Mesmo relacionamentos distantes serão
possivelmente detectados.
O Projeto de Comparação de Genomas: do que se trata?
Genes, genomas e dados genômicos
Os genes são as unidades hereditárias em todos os organismos viventes. Eles constituem componentes essenciais do genoma (o conjunto completo de informação genética) desses organismos, sendo responsáveis pelo desenvolvimento físico, pelo metabolismo e (até certo ponto) pelo comportamento desses organismos. A maioria dos genes codifica para proteínas, grandes moléculas feitas de longas cadeias de moléculas menores chamadas aminoácidos, respondendo pela maioria das reações bioquímicas desempenhadas pelas células. Apesar da maioria dos genes especificar a construção de proteínas, alguns produzem moléculas de RNA muito importantes;outros não codificam para nenhuma molécula mas são importantes de um ponto de vista regulatório ou estrutural. Em qualquer dos casos, as moléculas produzidas como resultado da atividade de um determinado gene são conhecidas como produtos gênicos.
Os genomas são moléculas de DNA ou RNA armazenadas e organizadas em um ou mais cromossomos, circulares ou lineares. Bactérias, por exemplo, armazenam seu genoma no citoplasma; organismos eucarióticos (seres com mais de uma célula, cada uma das quais possuindo um núcleo e outros compartimentos intracelulares bem desenvolvidos) organizam seu genoma no núcleo, assim como em organelas especializadas como as mitocôndrias e os cloroplastos (em plantas).
Desde os anos 90, esforços internacionais levaram a determinação do código genético completo de mais de 400 organismos (http://www.genomesonline.org/), como bactérias, leveduras, parasitas protozoários, plantas, invertebrados e vertebrados, incluindo o homem (Homo sapiens). Mais de 1500 investigações genômicas estão em andamento, representando organismos de interesse comercial, ambiental, industrial, ou importantes modelos de pesquisa. Com a continuidade desses trabalhos, novas seqüências genômicas estão tornando-se disponíveis em um ritmo cada vez mais acelerado, em adição a dados fragmentados de milhares de organismos, incluindo vírus. Os dados resultantes possuem o potencial de revelar os princípios básicos da genética, bioquímica e aspectos evolutivos desses organismos, assim como possibilitar o desenvolvimento de novos marcadores prognósticos, melhores medicamentos e vacinas, procedimentos diagnósticos aperfeiçoados, entre outros.
Distintas partes do genoma (de quase 100% em bactérias a aproximadamente 2% nos seres humanos) codificam para as proteínas, as quais por sua vez dirigem as atividades funcionais e estruturais das células. Análises computacionais podem predizer quais regiões do genoma codificam para as proteínas (cujo número varia de algumas centenas ou milhares em bactérias até 30.000 proteínas e suas variantes em humanos). Entretanto, a predição das funções celulares destas proteínas (estruturais, enzimas, transportadores, sinalizadores, etc.) é em sua maioria hipotética. A maior parte dessas possíveis funções foi atribuída por análises in silico (em computadores), usando as técnicas de comparação de seqüências com bancos de dados de proteínas. Entretanto, até o momento, somente uma pequena fração das proteínas preditas teve suas funções confirmadas por experimentos laboratoriais.
Genes codificantes para proteínas e sua anotação
A base de dados de genes e genomas RefSeq (Release 19 - September 2006 (http://www.ncbi.nlm.nih.gov/RefSeq/), registra mais de 2,8 milhões de genes codificantes preditos, de 3.774 organismos, incluindo vírus. Como dito acima, a maioria das identificações de genes supostamente codificando proteínas, assim como a própria seqüência protéica e sua anotação funcional (a adição de funções biológicas preditas e características estruturais ao dado bruto composto pela seqüência de DNA), foi feita com ferramentas computacionais e bancos de dados. Tal anotação funcional e estrutural foi aumentando com o passar dos anos, baseada na referência cruzada entre as crescentes bases de dados. Enquanto diversos esforços encontram-se em andamento para a construção de um conjunto referência bem verificado das proteínas, onde a função atribuída às mesmas tenha sido verificada experimentalmente, e adicionalmente utilize um conjunto referência de nomenclaturas para os genes, as proteínas e as funções celulares (como por exemplo o Gene Ontology – GO [http://www.geneontology.org]), assim como regras de anotação padronizadas, tal base de dados não existe até o momento.
O Projeto de Comparação de Genomas: melhorando a anotação
funcional das proteínas nas bases de dados
Com o passar dos anos, um grande corpo de informação secundária (estrutural, funcional, similaridades a outros registros e uma variedade de referências cruzadas) foi adicionada aos registros nas bases de dados de proteínas. Uma vez que tal informação seja adicionada, ela raramente sofre atualizações e/ou correções. Conseqüentemente, a anotacão da função protéica predita é freqüentemente incompleta, utiliza nomenclatura não-padronizada ou pode mesmo ser incorreta quando inferida de seqüências previamente anotadas de forma incorreta. Adicionalmente, muitas proteínas são compostas de vários domínios estruturais e/ou funcionais (módulos compreendendo unidades distintas do ponto de vista estrutural, funcional ou evolutivo), os quais podem ser ignorados por procedimentos de anotação automatizados. Além disso, a informação comparativa disponível atualmente é enorme quando comparada aos primeiros dias da genômica.
O objetivo principal do Projeto de Comparação de Genomas é a execução completa de comparações par-a-par entre todas as seqüências protéicas preditas, obtendo índices de similaridade que serão usados, juntamente com uma nomenclatura de genes e seus produtos padronizada (Gene Ontology - http://www.geneontology.org), na construção de um repositório de referência para a comunidade de anotadores, proporcionando uma fonte de dados inestimável para os biólogos. O programa de comparação de seqüências utilizado no Projeto de Comparação de Genomas é chamado SSEARCH (W.R. Pearson [1991] Genomics 11:635-650), uma implementação gratuita e aberta do algoritmo de Smith-Waterman (T. F. Smith and M. S. Waterman, [1981] J. Mol. Biol. 147:195-197), o qual encontra o melhor alinhamento local (do ponto de vista matemático) entre pares de seqüências.
Como resultado do projeto, uma anotação precisa e padronizada, a correção de inconsistências e a atribuição de possíveis funções à proteínas hipotéticas de função desconhecida serão facilitadas. Adicionalmente, proteínas com múltiplos domínios e elementos funcionais serão corretamente identificadas. Mesmo relacionamentos distantes serão possivelmente detectados.
Funções biológicas, sistemas complexos e biodiversidade
Os sistemas biológicos em uma célula são de grande complexidade, e nosso entendimento do conteúdo protéico total de uma célula, das interações entre as proteínas, das vias metabólicas e sua regulação é somente parcial (na melhor das hipóteses). Um banco de dados refletindo todos os relacionamentos entre as seqüências primárias das proteínas correspondentes de todos os organismos conhecidos ao nível genômico será de grande valor para a melhoria de nossa compreensão de toda essa complexidade.
Adicionalmente, a base de dados irá contribuir em vários aspectos experimentais na análise da biodiversidade em nosso planeta. Cientistas investigando amostras ambientais ou realizando análises fragmentadas de novos organismos serão capazes de utilizar os resultados do Projeto de Comparação de Genomas no estudo de diferentes aspectos da genética e bioquímica desses organismos. Além disso, a descrição e análise dos relacionamentos evolutivos entre as proteínas (e os organismos) baseados em tais análises genômicas representarão um grande passo no avanço de nosso conhecimento na evolução da estrutura dos genomas e na organização estrutural e bioquímica dos organismos. Iniciativas de grande escala como a Árvore da Vida (Tree of Life) e a catalogação da Biodiversidade irão se beneficiar em muito do Projeto de Comparação de Genomas e sua base de dados associada.
Novas drogas, vacinas e métodos diagnósticos
A pesquisa científica e o desenvolvimento (bio)tecnológico baseados na genômica estão fazendo um progresso crescente no desenvolvimento de novos métodos diagnósticos, assim como no desenvolvimento de novas drogas e vacinas. A genômica comparativa e o conhecimento das vias bioquímicas e processos celulares são de enorme importância nessa área. Por outro lado, análises funcionais e estudos sobre as interações entre as proteínas são de extrema importância para a compreensão de como os microrganismos, das células em organismos multicelulares e patógenos interagem com seu ambiente e seus hospedeiros, abrindo caminho para o desenho de novas estratégias de controle para doenças infecciosas e parasitárias, assim como de doenças metabólicas, crônicas ou degenerativas.
O World Community Grid e a anotação functional genômica
Comparações rigorosas par-a-par entre seqüências são
operações computacionalmente intensivas, e uma comparação
do tipo todos-contra-todos entre todas as proteínas preditas de todos
os genomas completamente seqüenciados é uma tarefa quase impossível
de ser atingida sem o suporte da grande estrutura em rede da Comunidade Mundial (World Community
Grid).
A matriz de informação resultante formará uma base de dados
de valor inestimável, a qual poderá ser continuamente incrementada
conforme novas seqüências genômicas se tornarem disponíveis,
formando o material básico para diversos estudos funcionais pela comunidade
científica.
* O que é o Projeto de Comparação de
Genomas, da Fiocruz?
* Por que o Projeto de Comparação de Genomas
compara seqüências protéicas?
* Como as proteínas são comparadas no Projeto
de Comparação de Genomas?
* Quais são os benefícios potenciais do Projeto
de Comparação de Genomas?
* Como posso participar do Projeto de Comparação
de Genomas?
* Como funciona o software do Projeto de Comparação
de Genomas?
* Quais computadores podem rodar o software?
* O que significam os círculos,
símbolos e letras no screen saver do Projeto de comparação
de Genomas?
O Projeto de Comparação de Genomas é uma iniciativa da Equipe de Bioinformática do Laboratório de Genômica Funcional e Bioinformática, pertencente ao Departamento de Bioquímica e Biologia Molecular do Instituto Oswaldo Cruz, Fiocruz, que utiliza técnicas de computação distribuída para contribuir seus recursos computacionais não-utilizados para calcular índices de similaridade entre todo o conteúdo protéico codificado nos genomas completos de centenas de organismos, incluindo o homem e diversas outras espécies de interesse médico, comercial, industrial ou de importância na pesquisa como organismos-modelo de estudos. Os índices de similaridade calculados serão utilizados, juntamente com nomenclatura padronizada (Gene Ontology), como um repositório de referência para a comunidade de anotadores, proporcionando uma fonte dados de enorme valor para os biólogos.
Somente uma fração do conteúdo protéico predito codificado em genomas completamente seqüenciados teve realmente sua função biológica e expressão confirmadas por experimentos laboratoriais. O processo de atribuição das funções biológicas preditas e características estruturais aos dados brutos (seqüência primaria da proteína) é chamado de anotação, o qual é cumprido principalmente pela sua comparação com as proteínas preditas armazenadas em diferentes bases de dados públicas em todo o mundo. Entretanto, esta anotação é freqüentemente incompleta, usa nomenclatura não-padronizada e pode ser incorreta quando inferida de seqüências previamente anotadas erroneamente. Conseqüentemente, um banco de dados comparativo do tipo todos-contra-todos será de grande importância como referência.
As seqüências biológicas (DNAs, RNAs e proteínas) são comparadas principalmente em pares por um processo denominado alinhamento par-a-par de seqüências, o qual consiste em posicionar as seqüências lado-a-lado de tal forma que o número de posições idênticas entre as duas seja maximizado. As seqüências podem ser alinhadas de forma global (contemplando toda a seqüência) ou local (enfatizando apenas partes da seqüência), dependendo do contexto e do propósito do alinhamento. O programa de comparação de seqüências utilizado no Projeto de Comparação de Genomas é chamado SSEARCH (W.R. Pearson [1991] Genomics 11:635-650), uma implementação gratuita e aberta do algoritmo de Smith-Waterman (T. F. Smith and M. S. Waterman, [1981] J. Mol. Biol. 147:195-197), o qual encontra o melhor alinhamento local (do ponto de vista matemático) entre pares de seqüências.
A base de dados comparativa resultante do projeto será de grande utilidade como referência para muitos projetos de pesquisa em aspectos funcionais, metabólicos, evolutivos, assim como uma fonte para a anotação correta de seqüências previamente obtidas assim como de novas seqüências genômicas;
Uma anotação precisa, a atribuição de possíveis funções à proteínas hipotéticas de função desconhecida, e a descrição dos relacionamentos evolutivos entre proteínas representará um grande passo rumo a uma melhor compreensão da organização genômica, sua evolução e as funções celulares;
A contribuição para o entendimento dos relacionamentos parasito-hospedeiro, e os meios para o desenvolvimento de novas drogas e vacinas, será de grande benefício para a comunidade científica como um todo;
Pesquisas em biodiversidade e novos organismos irão se beneficiar em muito com dados comparativos confiáveis;
Versões futuras serão incrementais, atualizando o
crescente banco de dados.
Tudo o que você precisa fazer para se unir ao Projeto de Comparação de Genomas é baixar e instalar o software livre oferecido pelo World Community Grid. Uma vez que o software tenha sido instalado, o seu computador irá automaticamente trabalhar quando você não estiver utilizando sua máquina, de forma que você possa continuar a usar seu computador da forma usual.
O software baixa automaticamente pequenas porções dos dados (seqüências protéicas preditas) e executa comparações entre elas para calcular de forma precisa os índices de similaridade entre as mesmas. Após o seu computador ter processado essas informações, os resultados serão enviados pelo World Community Grid para a Fiocruz, onde elas serão analisadas pela Equipe de Bioinformática do Laboratório de Genômica Funcional e Bioinformática, membro do Departamento de Bioquímica e Biologia Molecular do Instituto Oswaldo Cruz. A análise comparativa em larga escala utilizando o algoritmo de Smith-Waterman é computacionalmente intensiva, demandando grande poder computacional, razão pela qual o World Community Grid necessita que você (e seus amigos!) participem do Projeto de Comparação de Genomas.
No momento, os requisitos de hardware para este projeto são:
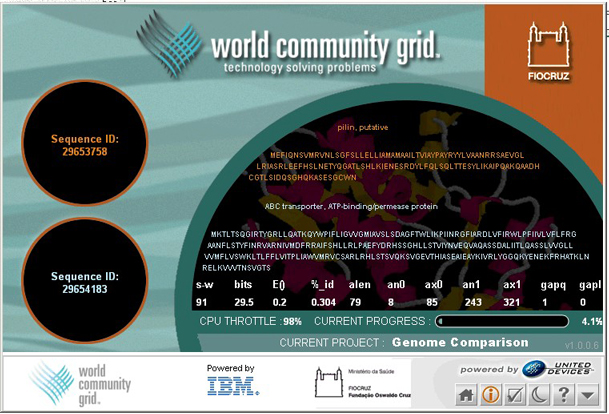 |
O painel apresentado em "Genome Comparison" representa as entidades envolvidas no processo de comparação e um sumário do resultado arquivado para um par destes.
Os círculos pequenos no lado esquerdo simbolizam dois diferentes genes, pertencentes a dois genomas distintos ou a um único genoma. Dentro de cada círculo podemos observar o número original que identifica a seqüência predita da proteína codificada pelo gene na base de dados.
O círculo grande no lado direito mostra as seqüências, sua descrições, e o nome abreviado das pontuações de similaridade e seus valores calculados para este par de sqüência particular.
As seqüências da proteína são representadas por letras ordenadas (como codificado em seus respectivos genes ). Cada uma destas letras representa um aminoácido diferente (M para o metionina, S para o serina, e assim por diante) na proteína.
A maioria das seqüências de proteína são hipotéticas ou putativas, o que significa que sua existência foi predita computacionalmente mas sua expressão pela célula ou organismo respectivos ainda não foram confirmados experimentalmente.
É possível infer um baixo nível de similaridade entre este par particular de seqüências, baseado nos valores arquivados para a pontuação de similaridade computada:
Parâmetro |
Valor |
Pequena descrição |
s-w |
[91] |
Smith-Waterman. A pontução obtida para o alinhamento de duas seqüências, de acordo com uma matriz de substituição particular |
bits |
[29.5] |
Bit score. Pontuação normalizada |
E() |
[0.2] |
Valor esperado ou E-value. Representa o número de alinhamentos com a mesma pontuação ou maior esperados por acaso. |
%_id |
[0.304] |
Fração ou posições identicas para um dado alinhamento. |
alen |
[79] |
Comprimento do alinhamento. |
an0 |
[8] |
Posição inicial da seqüência de consulta no alinhamento. |
ax0 |
[85] |
Posição final da seqüência de consulta no alinhamento. |
an1 |
[243] |
Start position of the subject sequence in the alignment |
ax1 |
[321] |
Posição inicial da seqüência de busca no alinhamento. |
gapq |
[1] |
Número de gaps introduzidos na seqüência de consulta durante o alinhamento. |
gapl |
[0] |
Número de gaps introduzidos na seqüência de busca durante o alinhamento. |
Pesquisadores
Wim Degrave, Ph.D
Antonio Basílio de Miranda, Ph.D
Estudantes de doutorado
Marcos Catanho, M.Sc
Thomas Dan Otto, M.Sc
Ana Carolina Ramos Guimarães, M.Sc
Colaboradores
Luiz Fernando Bessa Seibel, Ph.D - PUC, Rio de Janeiro, Brasil
José Antonio F. de Macêdo, Ph.D - PUC, Rio de Janeiro, Brasil
Melissa Lemos, Ph.D - PUC, Rio de Janeiro, Brasil
Sérgio Lifschitz, Ph.D - PUC, Rio de Janeiro, Brasil
Celso Ribeiro, Ph.D - PUC, Rio de Janeiro, Brasil
Fernando Alvarez Valin, Ph.D - Universidad de la República, Montevidéu, Uruguai
Hector Musto, Ph.D - Universidad de la República, Montevidéu, Uruguai