![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

This is the practical exercise for ClustalX. In this practical you will become familiar with performing alignments, changing parameters, performing profile alignments and assessing the quality of the alignments.ClustalX Practical
1. Load the sequences in the file "inf1.fas" into memory. Save the file to your home directory, or an appropriate location. There are 5 amino acid sequences in this file.
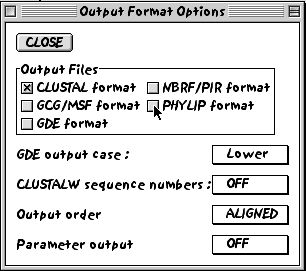
2. Under the alignment menu, choose the output format options and select the PHYLIP output format (we might wish to manipulate the alignment manually later, so this format is compatable with the SEAVIEW manual alignment software).
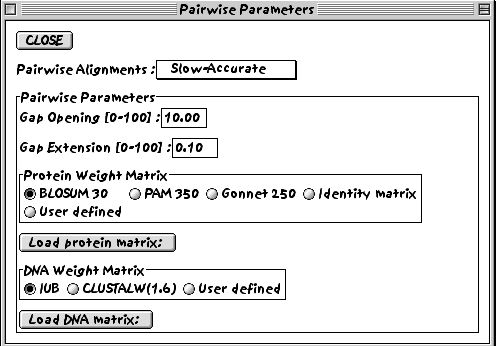
3. Look at the pairwise alignment parameters settings.
4. Look at the multiple alignment parameters settings.
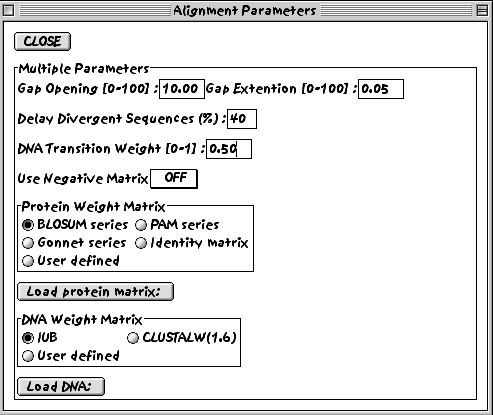
If we were dealing with a nucleotide sequence alignment, we could change the parameters for DNA sequences.
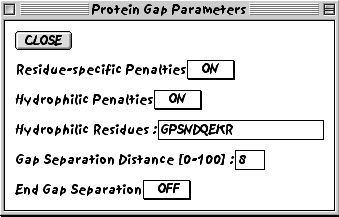
5. You can also decide to change the protein-specific gap parameters.
6. When you are satisfied with the alignment parameters, you should carry out a complete alignment.
7. Remove the gaps and realign with different penalties (either very small or very large values)
8. Remove the gaps and realign with different penalties (the opposite of whatever you chose at step 7).
9. Now we are going to perform a profile alignment. You must change the mode of operation of ClustalX from Multiple alignment to profile alignment.
10. When you switch to profile alignment mode, the screen will split and the bottom half of the screen is reserved to the second profile (note: you could have started the program in profile alignment mode and input 2 profiles from disk).
11. The file for performing the profile alignment is "inf2.fas". Save this file to your home directory.
12. Although there is the choice of aligning the second profile to the first, it is not a sensible option in most situations. In this case, the second profile is unaligned so this option makes very little sense. It is much more profitable to align the sequences in the second profile to the first profile. do this now.
13. You can lock the scroll bars together and look along the length of the alignment. If there are regions to be realigned, perhaps using a different scoring scheme then you can select those regions by switching back to multiple alignment mode, selecting the badly-aligned region and chosing the appropriate option in the alignments menu. Note: There might be a computer 'bug' in this section of the code.
14. You should now choose to ask the program to evaluate the alignment for regions of poor alignment. This is done under the Quality menu. I would suggest looking for low-scoring segments, initially looking for long stretehes (say 7 or more residues).
15. When you are satisfied that the alignment is not going to be improved by automated methods it might be appropriate to move to a manual alignment method in an effort to improve the alignment. You can now execute 'seaview' and load the PHYLIP-formatted output file from ClustalX.
16. Seaview is a manual alignment program, again using
the Vibrant library (which is part of the NCBI toolkit). It is available
for most computer platforms and can be retrieved from http://pbil.univ-lyon1.fr/.
The initial screen looks like this:
17. You can alter the properties of the program by choosing the props menu. One of the important things to change is the save format. The default is MASE, but the PHYLIP format is more useful.
If the program is configured to use ClustalW (text-version of ClustalX, it is possible to do some automated alignment. However, most of the features of automated alignment in seaview simply proceed by calling the Clustal program and it is probably a better idea to do any of these things directly in ClustalX. It is also possible to generate a consensus sequence.
18. You can define sets of sites and this will allow you to exclude regions of poor alignment. you can do this by creating a new set (remember to give this set a sensible name). Then you can select/deselect sites using the mouse.
19. When you are happy that only positions of unquestionable homology remain in the alignment, then you can save all of the sequences to disk. Note: it is also possible to just work on a subset of the sequences using the 'species' menu.

